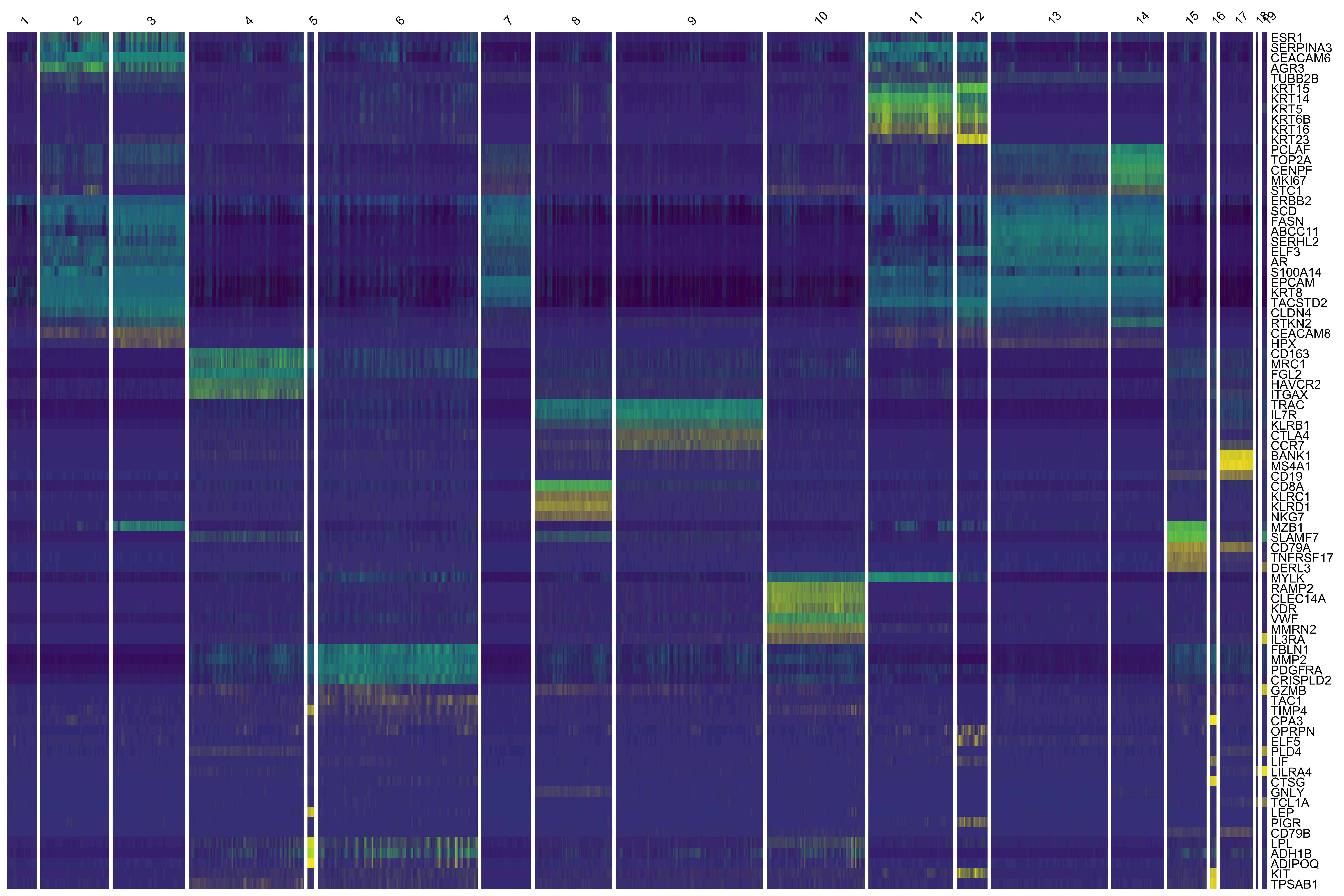
p_val avg_log2FC pct.1 pct.2 p_val_adj cluster gene
CPA3 0 7.343881 0.977 0.029 0 16 CPA3
CTSG 0 7.114698 0.878 0.011 0 16 CTSG
LILRA4.1 0 6.992717 0.939 0.015 0 19 LILRA4
ADIPOQ 0 6.860190 0.974 0.025 0 5 ADIPOQ
MS4A1 0 6.763083 0.919 0.027 0 17 MS4A1
BANK1 0 6.082192 0.889 0.037 0 17 BANK1